Welcome to Micro-C documentation

Overview
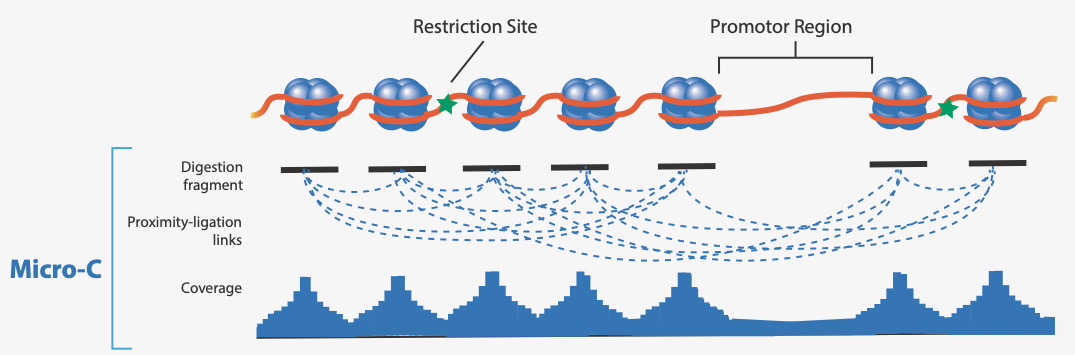
Dovetail™ Micro-C Kit uses the Micrococcal nuclease (MNase) enzyme instead of restriction enzymes for chromatin digestion, yielding 146 bp fragments distributed frequently across the genome.
Key benefits of Micro-C:
Sequence-independent chromatin fragmentation enables even genome-wide detection of chromatin contacts (up to 20% of the genome lacks coverage using restriction enzyme based Hi-C approaches)
Ultra-high nucleosome-level resolution of chromatin contacts
Highest signal-to-noise data with both enrichment of long-range informative reads and nucleosome protected fragments
The ability to detect higher-order features, such as chromatin loops, in proximity ligation data is dependent on enriching long-range informative reads to capture chromatin interaction frequency. The increased chromosome conformation informative reads combined with ultra-high-resolution improves loop calling compared to RE-based methods.
If you are using the Micro-C protocol as part of the Dovetail™ HiChIP MNase solution, please reffer to our HiChIP page for further instructions.
This guide will take you step by step on how to QC your Micro-C library, how to interparate the QC results and how to generate contact maps. If you don’t yet have a sequenced Micro-C library and you want to get familiar with the data, you can download Micro-C sequences libraries from our publicaly available data sets.
The QC process starts with aligning the reads to a reference genome then retaining high quality mapped reads. From there the mapped data will be used to generating a pairs file with pairtools, which categorizes pairs by read type and insert distance, this step both flags and removes PCR duplicates. Once pairs are categorized, counts of each class are summed and reported.
If this is your first time following this tutorial, please check the Before you begin page first.